

Performing longitudinal and paired sample comparisons with q2-longitudinal¶
Note
This guide assumes you have installed QIIME 2 using one of the procedures in the install documents.
This tutorial will demonstrate the various features of q2-longitudinal, a plugin that supports statistical and visual comparisons of longitudinal study designs and paired samples, to determine if/how samples change between observational “states”. “States” will most commonly be related to time or an environmental gradient, and for paired analyses (pairwise-distances and pairwise-differences) the sample pairs should typically consist of the same individual subject observed at two different time points. For example, patients in a clinical study whose stool samples are collected before and after receiving treatment.
“States” can also commonly be methodological, in which case sample pairs will usually be the same individual at the same time with two different methods. For example, q2-longitudinal could compare the effects of different collection methods, storage methods, DNA extraction methods, or any bioinformatic processing steps on the feature composition of individual samples.
Note
Many of the actions in q2-longitudinal take a metric value as input, which is usually a column name in a metadata file or a metadata-transformable artifact (including alpha diversity vectors, PCoA results, and many other QIIME 2 artifacts), or a feature ID in a feature table. The names of valid metric values in metadata files and metadata-transformable artifacts can be checked with the metadata tabulate command. Valid feature names (to use as metric values associated with a feature table) can be checked with the feature-data summarize command.
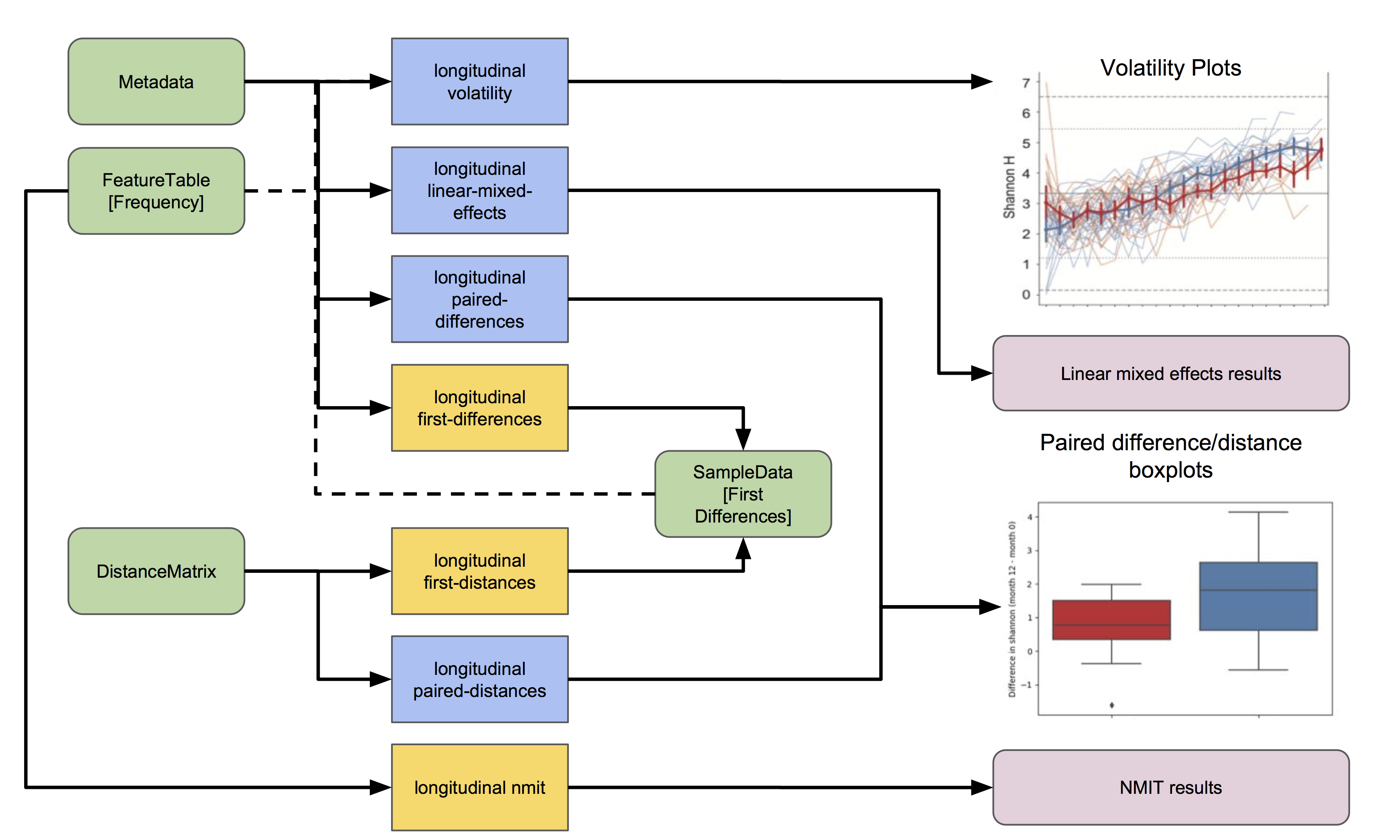
The following flowchart illustrates the workflow involved in all q2-longitudinal analyses (figure key). Each of these actions is described in more detail in the tutorials below.
In the examples below, we use data from the ECAM study, a longitudinal study of infants’ and mothers’ microbiota from birth through 2 years of life. First let’s create a new directory and download the relevant tutorial data.
mkdir longitudinal-tutorial
cd longitudinal-tutorial
Download URL: https://data.qiime2.org/2024.5/tutorials/longitudinal/sample_metadata.tsv
Save as: ecam-sample-metadata.tsv
wget \
-O "ecam-sample-metadata.tsv" \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/sample_metadata.tsv"
curl -sL \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/sample_metadata.tsv" > \
"ecam-sample-metadata.tsv"
Download URL: https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_shannon.qza
Save as: shannon.qza
wget \
-O "shannon.qza" \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_shannon.qza"
curl -sL \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_shannon.qza" > \
"shannon.qza"
Download URL: https://data.qiime2.org/2024.5/tutorials/longitudinal/unweighted_unifrac_distance_matrix.qza
Save as: unweighted_unifrac_distance_matrix.qza
wget \
-O "unweighted_unifrac_distance_matrix.qza" \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/unweighted_unifrac_distance_matrix.qza"
curl -sL \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/unweighted_unifrac_distance_matrix.qza" > \
"unweighted_unifrac_distance_matrix.qza"
Pairwise difference comparisons¶
Pairwise difference tests determine whether the value of a specific metric changed significantly between pairs of paired samples (e.g., pre- and post-treatment).
This visualizer currently supports comparison of feature abundance (e.g., microbial sequence variants or taxa) in a feature table, or of metadata values in a sample metadata file. Alpha diversity values (e.g., observed sequence variants) and beta diversity values (e.g., principal coordinates) are useful metrics for comparison with these tests, and should be contained in one of the metadata files given as input. In the example below, we will test whether alpha diversity (Shannon diversity index) changed significantly between two different time points in the ECAM data according to delivery mode.
qiime longitudinal pairwise-differences \
--m-metadata-file ecam-sample-metadata.tsv \
--m-metadata-file shannon.qza \
--p-metric shannon \
--p-group-column delivery \
--p-state-column month \
--p-state-1 0 \
--p-state-2 12 \
--p-individual-id-column studyid \
--p-replicate-handling random \
--o-visualization pairwise-differences.qzv
Pairwise distance comparisons¶
The pairwise-distances visualizer also assesses changes between paired samples from two different “states”, but instead of taking a metadata column or artifact as input, it operates on a distance matrix to assess the distance between “pre” and “post” sample pairs, and tests whether these paired differences are significantly different between different groups, as specified by the group-column parameter. Here we use this action to compare the stability of the microbiota compositions of vaginally born and cesarean-delivered infants over a 12-month time frame in the ECAM data set.
qiime longitudinal pairwise-distances \
--i-distance-matrix unweighted_unifrac_distance_matrix.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--p-group-column delivery \
--p-state-column month \
--p-state-1 0 \
--p-state-2 12 \
--p-individual-id-column studyid \
--p-replicate-handling random \
--o-visualization pairwise-distances.qzv
Linear mixed effect models¶
Linear mixed effects (LME) models test the relationship between a single response variable and one or more independent variables, where observations are made across dependent samples, e.g., in repeated-measures sampling experiments. This implementation takes at least one numeric state-column (e.g., Time) and one or more comma-separated group-columns (which may be categorical or numeric metadata columns; these are the fixed effects) as independent variables in a LME model, and plots regression plots of the response variable (“metric”) as a function of the state column and each group column. Additionally, the individual-id-column parameter should be a metadata column that indicates the individual subject/site that was sampled repeatedly. The response variable may either be a sample metadata mapping file column or a feature ID in the feature table. A comma-separated list of random effects can also be input to this action; a random intercept for each individual is included by default, but another common random effect that users may wish to use is a random slope for each individual, which can be set by using the state-column value as input to the random-effects parameter. Here we use LME to test whether alpha diversity (Shannon diversity index) changed over time and in response to delivery mode, diet, and sex in the ECAM data set.
Note
Deciding whether a factor is a fixed effect or a random effect can be complicated. In general, a factor should be a fixed effect if the different factor levels (metadata column values) represent (more or less) all possible discrete values. For example, delivery mode, sex, and diet (dominantly breast-fed or formula-fed) are designated as fixed effects in the example below. Conversely, a factor should be a random effect if its values represent random samples from a population. For example, we could imagine having metadata variables like body-weight, daily-kcal-from-breastmilk, number-of-peanuts-eaten-per-day, or mg-of-penicillin-administered-daily; such values would represent random samples from within a population, and are unlikely to capture all possible values representative of the whole population. Not sure about the factors in your experiment? 🤔 Consult a statistician or reputable statistical tome for guidance. 📚
qiime longitudinal linear-mixed-effects \
--m-metadata-file ecam-sample-metadata.tsv \
--m-metadata-file shannon.qza \
--p-metric shannon \
--p-group-columns delivery,diet,sex \
--p-state-column month \
--p-individual-id-column studyid \
--o-visualization linear-mixed-effects.qzv
The visualizer produced by this command contains several results. First, the input parameters are shown at the top of the visualization for convenience (e.g., when flipping through multiple visualizations it is useful to have a summary). Next, the “model summary” shows some descriptive information about the LME model that was trained. This just shows descriptive information about the “groups”; in this case, groups will be individuals (as set by the --p-individual-id-column). The main results to examine will be the “model results” below the “model summary”. These results summarize the effects of each fixed effect (and their interactions) on the dependent variable (shannon diversity). This table shows parameter estimates, estimate standard errors, z scores, P values (P>|z|), and 95% confidence interval upper and lower bounds for each parameter. We see in this table that shannon diversity is significantly impacted by month of life and by diet, as well as several interacting factors. More information about LME models and the interpretation of these data can be found on the statsmodels LME description page, which provides a number of useful technical references for further reading.
Finally, scatter plots categorized by each “group column” are shown at the bottom of the visualization, with linear regression lines (plus 95% confidence interval in grey) for each group. If --p-lowess is enabled, instead locally weighted averages are shown for each group. Two different groups of scatter plots are shown. First, regression scatterplots show the relationship between state_column (x-axis) and metric (y-axis) for each sample. These plots are just used as a quick summary for reference; users are recommended to use the volatility visualizer for interactive plotting of their longitudinal data. Volatility plots can be used to qualitatively identify outliers that disproportionately drive the variance within individuals and groups, including by inspecting residuals in relation to control limits (see note below and the section on “Volatility analysis” for more details).
The second set of scatterplots are fit vs. residual plots, which show the relationship between metric predictions for each sample (on the x-axis), and the residual or observation error (prediction - actual value) for each sample (on the y-axis). Residuals should be roughly zero-centered and normal across the range of measured values. Uncentered, systematically high or low, and autocorrelated values could indicate a poor model. If your residual plots look like an ugly mess without any apparent relationship between values, you are doing a good job. If you see a U-shaped curve or other non-random distribution, either your predictor variables (group_columns and/or random_effects) are failing to capture all explanatory information, causing information to leak into your residuals, or else you are not using an appropriate model for your data 🙁. Check your predictor variables and available metadata columns to make sure you aren’t missing anything.
Note
If you want to dot your i’s and cross your t’s, residual and predicted values for each sample can be obtained in the “Download raw data as tsv” link below the regression scatterplots. This file can be input as metadata to the volatility visualizer to check whether residuals are correlated with other metadata columns. If they are, those columns should probably be used as prediction variables in your model! Control limits (± 2 and 3 standard deviations) can be toggled on/off to easily identify outliers, which can be particularly useful for re-examining fit vs. residual plots with this visualizer. 🍝
Volatility analysis¶
The volatility visualizer generates interactive line plots that allow us to assess how volatile a dependent variable is over a continuous, independent variable (e.g., time) in one or more groups. Multiple metadata files (including alpha and beta diversity artifacts) and FeatureTable[RelativeFrequency] tables can be used as input, and in the interactive visualization we can select different dependent variables to plot on the y-axis.
Here we examine how variance in Shannon diversity and other metadata changes across time (set with the state-column parameter) in the ECAM cohort, both in groups of samples (interactively selected as described below) and in individual subjects (set with the individual-id-column parameter).
qiime longitudinal volatility \
--m-metadata-file ecam-sample-metadata.tsv \
--m-metadata-file shannon.qza \
--p-default-metric shannon \
--p-default-group-column delivery \
--p-state-column month \
--p-individual-id-column studyid \
--o-visualization volatility.qzv
In the resulting visualization, a line plot appears on the left-hand side of the plot and a panel of “Plot Controls” appears to the right. These “Plot Controls” interactively adjust several variables and parameters. This allows us to determine how groups’ and individuals’ values change across a single independent variable, state-column. Interective features in this visualization include:
The “Metric column” tab lets us select which continuous metadata values to plot on the y-axis. All continuous numeric columns found in metadata/artifacts input to this action will appear as options in this drop-down tab. In this example, the initial variable plotted in the visualization is shannon diversity because this column was designated by the optional
default-metricparameter.The “Group column” tab lets us select which categorical metadata values to use for calculating mean values. All categorical metadata columns found in metadata/artifacts input to this action will appear as options in this drop-down tab. These mean values are plotted on the line plot, and the thickness and opacity of these mean lines can be modified using the slider bars in the “Plot Controls” on the right-hand side of the visualization. Error bars (standard deviation) can be toggled on and off with a button in the “Plot Controls”.
Longitudinal values for each individual subject are plotted as “spaghetti” lines (so-called because this tangled mass of individual vectors looks like a ball of spaghetti). The thickness and opacity of spaghetti can be modified using the slider bars in the “Plot Controls” on the right-hand side of the visualization.
Color scheme can be adjusted using the “Color scheme” tab.
Global mean and warning/control limits (2X and 3X standard deviations from global mean) can be toggled on/off with the buttons in the “Plot Controls”. The goal of plotting these values is to show how a variable is changing over time (or a gradient) in relation to the mean. Large departures from the mean values can cross the warning/control limits, indicating a major disruption at that state; for example, antibiotic use or other disturbances impacting diversity could be tracked with these plots.
Group mean lines and spaghetti can also be modified with the “scatter size” and “scatter opacity” slider bars in the “Plot Controls”. These adjust the size and opacity of individual points. Maximize scatter opacity and minimize line opacity to transform these into longitudinal scatter plots!
Relevant sample metadata at individual points can be viewed by hovering the mouse over a point of interest.
If the interactive features of this visualization don’t quite scratch your itch, you can use the external vega editor to edit the plot. Click on the “View Source” button then copy the contents of that page and paste them into the left column of this webpage https://vega.github.io/editor.
Buon appetito! 🍝
First differencing to track rate of change¶
Another way to view time series data is by assessing how the rate of change differs over time. We can do this through calculating first differences, which is the magnitude of change between successive time points. If \(Y_\text{t}\) is the value of metric \(Y\) at time \(t\), the first difference at time \(t\), \({\Delta}Y_\text{t} = Y_\text{t} - Y_\text{t-1}\). This calculation is performed at fixed intervals, so for each interval \({\Delta}Y_\text{t}\) is not calculated for subjects that are missing samples at times \(t\) or \(t-1\). This transformation is performed in the first-differences method in q2-longitudinal.
qiime longitudinal first-differences \
--m-metadata-file ecam-sample-metadata.tsv \
--m-metadata-file shannon.qza \
--p-state-column month \
--p-metric shannon \
--p-individual-id-column studyid \
--p-replicate-handling random \
--o-first-differences shannon-first-differences.qza
This outputs a SampleData[FirstDifferences] artifact, which can then be viewed, e.g., with the volatility visualizer or analyzed with linear-mixed-effects or other methods.
A similar method is first-distances, which instead identifies the beta diversity distances between successive samples from the same subject. The pairwise distance between all samples can already be calculated by the beta or core-metrics methods in q2-diversity, so this method simply identifies the distances between successive samples collected from the same subject and outputs this series of values as metadata that can be consumed by other methods.
qiime longitudinal first-distances \
--i-distance-matrix unweighted_unifrac_distance_matrix.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--p-state-column month \
--p-individual-id-column studyid \
--p-replicate-handling random \
--o-first-distances first-distances.qza
This output can be used in the same way as the output of first-differences. The output of first-distances is particularly empowering, though, because it allows us to analyze longitudinal changes in beta diversity using actions that cannot operate directly on a distance matrix, such as linear-mixed-effects.
qiime longitudinal linear-mixed-effects \
--m-metadata-file first-distances.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--p-metric Distance \
--p-state-column month \
--p-individual-id-column studyid \
--p-group-columns delivery,diet \
--o-visualization first-distances-LME.qzv
Tracking rate of change from static timepoints¶
The first-differences and first-distances methods both have an optional “baseline” parameter to instead calculate differences from a static point (e.g., baseline or a time point when a treatment is administered: \({\Delta}Y_\text{t} = Y_\text{t} - Y_\text{0}\)). Calculating baseline differences can help tease apart noisy longitudinal data to reveal underlying trends in individual subjects or highlight significant experimental factors related to changes in diversity or other dependent variables.
qiime longitudinal first-distances \
--i-distance-matrix unweighted_unifrac_distance_matrix.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--p-state-column month \
--p-individual-id-column studyid \
--p-replicate-handling random \
--p-baseline 0 \
--o-first-distances first-distances-baseline-0.qza
Note
Fun fact! We can also use the first-distances method to track longitudinal change in the proportion of features that are shared between an individual’s samples. This can be performed by calculating pairwise Jaccard distance (proportion of features that are not shared) between each pair of samples and using this as input to first-distances. This is particularly useful for pairing with the baseline parameter, e.g., to determine how unique features are lost/gained over the course of an experiment.
Non-parametric microbial interdependence test (NMIT)¶
Within microbial communities, microbial populations do not exist in isolation but instead form complex ecological interaction webs. Whether these interdependence networks display the same temporal characteristics within subjects from the same group may indicate divergent temporal trajectories. NMIT evaluates how interdependencies of features (e.g., microbial taxa, sequence variants, or OTUs) within a community might differ over time between sample groups. NMIT performs a nonparametric microbial interdependence test to determine longitudinal sample similarity as a function of temporal microbial composition. For each subject, NMIT computes pairwise correlations between each pair of features. Between-subject distances are then computed based on a distance norm between each subject’s microbial interdependence correlation matrix. For more details and citation, please see Zhang et al., 2017.
Note
NMIT, as with most longitudinal methods, largely depends on the quality of the input data. This method will only work for longitudinal data (i.e., the same subjects are sampled repeatedly over time). To make the method robust, we suggest a minimum of 5-6 samples (time points) per subject, but the more the merrier. NMIT does not require that samples are collected at identical time points (and hence is robust to missing samples) but this may impact data quality if highly undersampled subjects are included, or if subjects’ sampling times do not overlap in biologically meaningful ways. It is up to the users to ensure that their data are high quality and the methods are used in a biologically relevant fashion.
Note
NMIT can take a long time to run on very large feature tables. Removing low-abundance features and collapsing feature tables on taxonomy (e.g., to genus level) will improve runtime.
First let’s download a feature table to test. Here we will test genus-level taxa that exhibit a relative abundance > 0.1% in more than 15% of the total samples.
Download URL: https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_table_taxa.qza
Save as: ecam-table-taxa.qza
wget \
-O "ecam-table-taxa.qza" \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_table_taxa.qza"
curl -sL \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_table_taxa.qza" > \
"ecam-table-taxa.qza"
Now we are ready run NMIT. The output of this command is a distance matrix that we can pass to other QIIME2 commands for significance testing and visualization.
qiime longitudinal nmit \
--i-table ecam-table-taxa.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--p-individual-id-column studyid \
--p-corr-method pearson \
--o-distance-matrix nmit-dm.qza
Now let’s put that distance matrix to work. First we will perform PERMANOVA tests to evaluate whether between-group distances are larger than within-group distance.
Note
NMIT computes between-subject distances across all time points, so each subject (as defined the --p-individual-id-column parameter used above) gets compressed into a single “sample” representing that subject’s longitudinal microbial interdependence. This new “sample” will be labeled with the SampleID of one of the subjects with a matching individual-id; this is done for the convenience of passing this distance matrix to downstream steps without needing to generate a new sample metadata file but it means that you must pay attention. For significance testing and visualization, only use group columns that are uniform across each individual-id. DO NOT ATTEMPT TO USE METADATA COLUMNS THAT VARY OVER TIME OR BAD THINGS WILL HAPPEN. For example, in the tutorial metadata a patient is labeled antiexposedall==y only after antibiotics have been used; this is a column that you should not use, as it varies over time. Now have fun and be responsible.
qiime diversity beta-group-significance \
--i-distance-matrix nmit-dm.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--m-metadata-column delivery \
--o-visualization nmit.qzv
Finally, we can compute principal coordinates and use Emperor to visualize similarities among subjects (not individual samples; see the note above).
qiime diversity pcoa \
--i-distance-matrix nmit-dm.qza \
--o-pcoa nmit-pc.qza
qiime emperor plot \
--i-pcoa nmit-pc.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--o-visualization nmit-emperor.qzv
So there it is. We can use PERMANOVA test or other distance-based statistical tests to determine whether groups exhibit different longitudinal microbial interdependence relationships, and PCoA/emperor to visualize the relationships among groups of subjects. Don’t forget the caveats mentioned above about using and interpreting NMIT. Now be safe and have fun.
Feature volatility analysis¶
Note
This pipeline is a supervised regression method. Read the sample classifier tutorial for more details on the general process, outputs (e.g., feature importance scores), and interpretation of supervised regression models.
This pipeline identifies features that are predictive of a numeric metadata column, “state_column” (e.g., time), and plots their relative frequencies across states using interactive feature volatility plots (only important features are plotted). A supervised learning regressor is used to identify important features and assess their ability to predict sample states. state_column will typically be a measure of time, but any numeric metadata column can be used and this is not strictly a longitudinal method, unless if the individual_id_column parameter is used (in which case feature volatility plots will contain per-individual spaghetti lines, as described above). 🍝
Let’s test this out on the ECAM dataset. First download a table to work with:
Download URL: https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_table_maturity.qza
Save as: ecam-table.qza
wget \
-O "ecam-table.qza" \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_table_maturity.qza"
curl -sL \
"https://data.qiime2.org/2024.5/tutorials/longitudinal/ecam_table_maturity.qza" > \
"ecam-table.qza"
qiime longitudinal feature-volatility \
--i-table ecam-table.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--p-state-column month \
--p-individual-id-column studyid \
--p-n-estimators 10 \
--p-random-state 17 \
--output-dir ecam-feat-volatility
Output artifacts:
Output visualizations:
All of the parameters used in this pipeline are described for other q2-longitudinal actions or in the sample classifier tutorial, so will not be discussed here. This pipeline produces multiple outputs:
volatility-plotcontains an interactive feature volatility plot. This is very similar to the plots produced by thevolatilityvisualizer described above, with a couple key differences. First, only features are viewable as “metrics” (plotted on the y-axis). Second, feature metadata (feature importances and descriptive statistics) are plotted as bar charts below the volatility plot. The relative frequencies of different features can be plotted in the volatility chart by either selecting the “metric” selection tool, or by clicking on one of the bars in the bar plot. This makes it convenient to select features for viewing based on importance or other feature metadata. By default, the most important feature is plotted in the volatility plot when the visualization is viewed. Different feature metadata can be selected and sorted using the control panel to the right of the bar charts. Most of these should be self-explanatory, except for “cumulative average change” (the cumulative magnitude of change, both positive and negative, across states, and averaged across samples at each state), and “net average change” (positive and negative “cumulative average change” is summed to determine whether a feature increased or decreased in abundance between baseline and end of study).accuracy-resultsdisplay the predictive accuracy of the regression model. This is important to view, as important features are meaningless if the model is inaccurate. See the sample classifier tutorial for more description of regressor accuracy results.feature-importancecontains the importance scores of all features. This is viewable in the feature volatility plot, but this artifact is nonetheless output for convenience. See the sample classifier tutorial for more description of feature importance scores.filtered-tableis aFeatureTable[RelativeFrequency]artifact containing only important features. This is output for convenience.sample-estimatorcontains the trained sample regressor. This is output for convenience, just in case you plan to regress additional samples. See the sample classifier tutorial for more description of theSampleEstimatortype.
Now we will cover basic interpretation of these data. By looking at the accuracy-results, we see that the regressor model is actually quite accurate, even though only 10 estimators were used for training the regressor (in practice a larger number of estimators should be used, and the default for the --p-n-estimators parameter is 100 estimators; see the sample classifier tutorial for more description of this parameter). Great! The feature importances will be meaningful. Month of life can be accurately predicted based on the ASV composition of these samples, suggesting that a programmatic succession of ASVs occurs during early life in this childhood cohort.
Next we will view the feature volatility plot. We see that the most important feature is more than twice as important as the second most important feature, so this one is really predictive of a subject’s age! Sure enough, looking at the volatility chart we see that this feature is almost entirely absent at birth in most subjects, but increases gradually starting at around 8 months of life. Its average frequency is greater in vaginally born subjects than cesarean-delivered subjects, so could be an interesting candidate for statistical testing, e.g., with linear-mixed-effects. We can also use metadata tabulate to merge the feature importance data with taxonomy assignments to determine the taxonomic classification of this ASV (and other important features).
“Maturity Index” prediction¶
Note
This analysis currently works best for comparing groups that are sampled fairly evenly across time (the column used for regression). Datasets that contain groups sampled sporadically at different times are not supported, and users should either filter out those samples or “bin” them with other groups prior to using this visualizer.
Note
This analysis will only work on data sets with a large sample size, particularly in the “control” group, and with sufficient biological replication at each time point.
Note
This pipeline is a supervised regression method. Read the sample classifier tutorial for more details on the general process, outputs (e.g., feature importance scores), and interpretation of supervised regression models.
This method calculates a “microbial maturity” index from a regression model trained on feature data to predict a given continuous metadata column (“state_column”), e.g., to predict a subject’s age as a function of microbiota composition. This method is different from standard supervised regression because it quantifies the relative rate of change over time in two or more groups. The model is trained on a subset of control group samples, then predicts the column value for all samples. This visualization computes maturity index z-scores (MAZ) to compare relative “maturity” between each group, as described in Sathish et al. 2014. This method was originally designed to predict between-group differences in intestinal microbiome development by age, so state_column should typically be a measure of time. Other types of continuous metadata gradients might be testable, as long as two or more different “treatment” groups are being compared with a large number of biological replicates in the “control” group and treatment groups are sampled at the same “states” (time or position on gradient) for comparison. However, we do not necessarily recommend or offer technical support for unusual approaches.
Here we will compare microbial maturity between vaginally born and cesarean-delivered infants as a function of age in the ECAM dataset.
qiime longitudinal maturity-index \
--i-table ecam-table.qza \
--m-metadata-file ecam-sample-metadata.tsv \
--p-state-column month \
--p-group-by delivery \
--p-individual-id-column studyid \
--p-control Vaginal \
--p-test-size 0.4 \
--p-stratify \
--p-random-state 1010101 \
--output-dir maturity
Output artifacts:
Output visualizations:
This pipeline produces several output files:
accuracy_results.qzvcontains a linear regression plot of predicted vs. expected values on all control test samples (as described in the sample classifier tutorial). This is a subset of “control” samples that were not used for model training (the fraction defined by thetest-sizeparameter).volatility-plots.qzvcontains an interactive volitility chart. This visualization can be useful for assessing how MAZ and other metrics change over time in each sample group (by default, thegroup_bycolumn is used but other sample metadata may be selected for grouping samples). The default metric displayed on this chart is MAZ scores for the chosenstate_column. The “prediction” (predicted “state_column” values) and state_column “maturity” metrics are other metrics calculated by this plugin that can be interesting to explore. See Sathish et al. 2014 for more details on the MAZ and maturity metrics.clustermap.qzvcontains a heatmap showing the frequency of each important feature across time in each group. This plot is useful for visualizing how the frequency of important features changes over time in each group, demonstrating how different patterns of feature abundance (e.g., trajectories of development in the case of age or time-based models) may affect model predictions and MAZ scores. Important features shown along the x-axis; samples grouped and ordered bygroup_byandstate_columnare shown on the y-axis. See heatmap for details on how features are clustered along the x-axis (default parameters are used).maz_scores.qzacontains MAZ scores for each sample (excluding training samples). This is useful for downstream testing as described below.predictions.qzacontains “state column” predictions for each sample (excluding training samples). These predictions are used to calculate the MAZ scores, and a subset (control test samples) are used to assess model accuracy. Nonetheless, the predictions are supplied in case they prove useful… These are also available for viewing in the volatility plots.feature_importance.qzacontains importance scores for all features included in the final regression model. If theoptimize-feature-selectionparameter is used, this will only contain important features; if not, importance scores will be assigned to all features in the original feature table.sample_estimator.qzacontains the trainedSampleEstimator[Regressor]. You probably will not want to re-use this estimator for predicting other samples (since it is trained on a subset of samples), but nevertheless it is supplied for the curious and intrepid.model_summary.qzvcontains a summary of the model parameters used by the supervised learning estimator, as described in the sample classifier tutorial for the equivalently named outputs from theclassify-samplespipeline.
So what does this all show us? In the ECAM dataset that we are testing here, we see that MAZ scores are suppressed in Cesarean-delivered subjects in the second year of life, compared to vaginally born subjects (See volatility-plots.qzv). Several important sequence variants exhibit reduced frequency during this time frame, suggesting involvement in delayed maturation of the Cesarean cohort (See clustermap.qzv).
Note that none of the results presented so far actually confirm a statistical difference between groups. Want to take this analysis to the next level (with multivariate statistical testing)? Use the MAZ scores (or possibly predictions) as input metrics (dependent variables) in linear mixed effects models (as described above).